ポイント
- 患者の生存に影響する細胞を、1細胞単位で推定できる新しい解析手法scSurvを開発し、単一細胞レベルで、どの細胞が予後に有利または不利に関与するかを定量的に評価
- 深層生成モデルとCox比例ハザードモデルを統合し、単一細胞解析と生存解析を直接結びつけ、単一細胞データと臨床生存データを同時に扱う解析が可能に
- 予後を左右する細胞集団や関連遺伝子を特定し、治療標的やバイオマーカー探索への応用が期待され、既存のバルクRNAシーケンスと臨床データを活用した、個別化医療の基盤に
概要
東京科学大学(Science Tokyo)総合研究院 難治疾患研究所 計算システム生物学分野の水越周良大学院生、島村徹平教授、ならびに国立がん研究センター研究所計算生命科学ユニットの小嶋泰弘独立ユニット長(東京科学大学 総合研究院 難治疾患研究所 計算システム生物学分野 連携研究員)らの研究グループは、予後に影響する細胞を一細胞解像度で推定する新しい情報解析手法「scSurv」を開発しました。
近年、単一細胞解析の進展により、がん組織内に存在する多様な細胞状態(不均一性)を詳細に捉えることが可能になってきました。一方で、こうした不均一性が患者の予後にどのように関与しているのかを、単一細胞レベルで直接評価することは容易ではなく、単一細胞データと臨床生存データを結び付ける解析基盤の整備が求められていました。
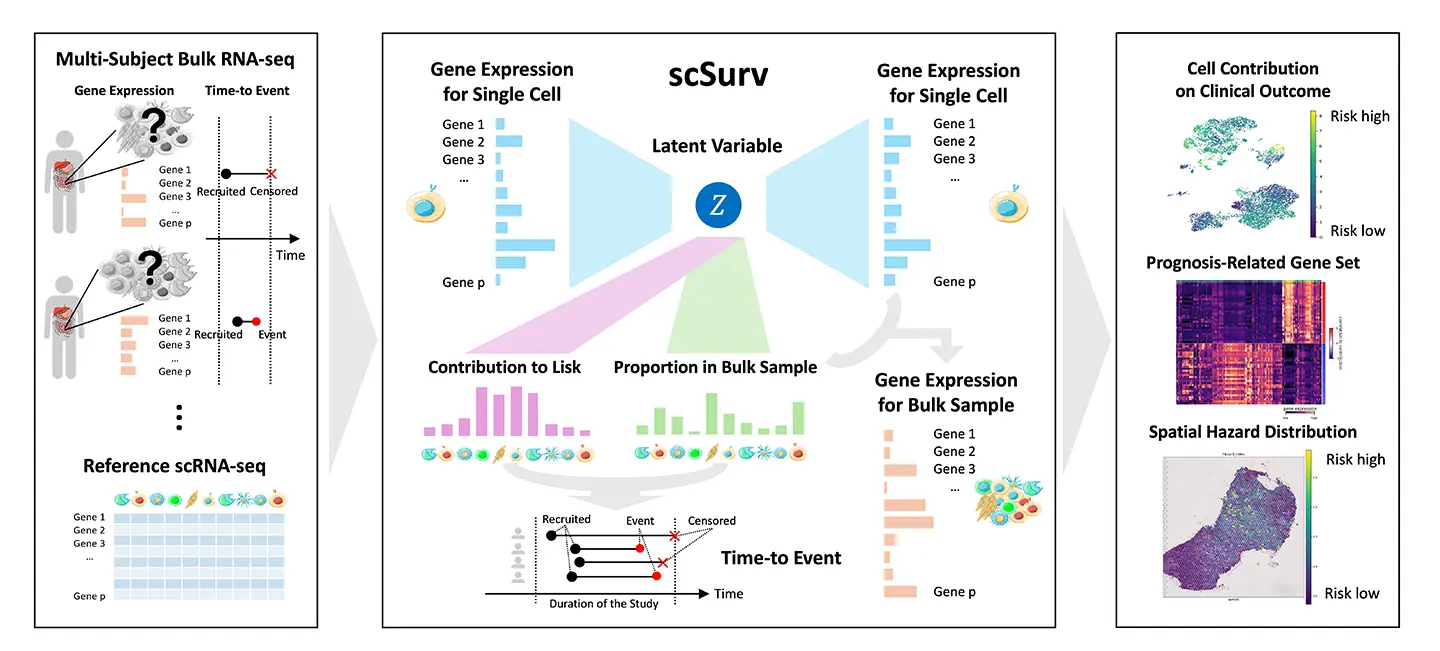
そこで本研究グループは、単一細胞RNAシーケンス[用語1]データの深層生成モデル[用語2]と、生存解析で広く用いられている Cox比例ハザードモデル[用語3]を統合することで、各細胞が臨床転帰(生存)に与える寄与を定量化できる新しい手法「scSurv」を開発しました。scSurvにより、予後に有利または不利に関与する細胞群の同定に加え、それらの寄与度と関連する遺伝子の抽出も可能になります。
本手法は、シミュレーションデータおよび実データを用いて検証され、メラノーマの解析においては、既知の予後関連マクロファージを同定できることが示されました。さらに、腎細胞がんの解析では、空間トランスクリプトーム解析[用語4]と組み合わせることで、組織切片上におけるハザード分布を推定できることも示されました。加えて、複数のがん種に共通する予後関連遺伝子の同定や、感染症データへの適用可能性も示されており、臨床転帰解析の高度化や治療標的探索への貢献が期待されます。
本成果は、国際学術誌「Bioinformatics 」オンライン版において、2025年12月22日に公開されました。
背景
がん組織は多様な細胞から構成されており、個々の細胞が持つ性質の違い(不均一性)が、がんの進行や治療効果に大きな影響を与えることが知られています。近年、単一細胞トランスクリプトーム解析の発展により、細胞ごとの遺伝子発現を網羅的に解析することが可能になりました。しかし、この技術はコストが高く、数千人規模の患者集団(コホート)に対して適用し、生存期間などの臨床情報と結び付けて解析することは、経済的・技術的に困難でした。
一方、従来のバルクRNAシーケンス[用語5]については、これまでに蓄積された膨大な臨床情報付きデータセット(TCGAなど)が存在します。しかし、従来の解析手法では、バルクデータから細胞タイプレベルの構成比率を推定することはできても、個々の細胞の状態の違い、すなわち細胞レベルの解像度にまで踏み込み、それらが予後にどのように影響するのかを解析することはできませんでした。
そのため、既存のビッグデータを活用しつつ、単一細胞レベルの解像度で疾患の予後予測や病態メカニズムの解明を可能とする、新たな解析手法の開発が求められていました。
研究成果
本研究グループは、単一細胞RNAシーケンスデータを参照しながら、臨床情報付きのバルクRNAシーケンスデータを単一細胞レベルに分解し、さらにその結果を生存解析に統合する深層学習モデル「scSurv」を開発しました。本研究の主な成果は以下の通りです。
-
個々の細胞の予後への寄与度を定量化
scSurvは、細胞の種類(細胞タイプ)ごとの解析にとどまらず、個々の細胞が持つ微細な状態の違いを考慮することで、各単一細胞が患者の生存に与える寄与度を推定することに成功しました。シミュレーションデータを用いた検証では、既存の細胞タイプレベルの解像度に基づく手法よりも高精度に、予後への寄与を推定できることが実証されました。 -
予後に関連する重要細胞・遺伝子の同定
メラノーマ(悪性黒色腫)のデータ解析において、scSurvは予後を悪化させる特定のマクロファージ集団を同定しました。さらに、その細胞群の特徴として、SPP1遺伝子の高発現が予後不良と、TNFSF10遺伝子の高発現が予後良好と関連することを見出しました。これらの結果は既知の生物学的知見と一致しており、本手法の信頼性が確認されました。 -
空間的ハザードマップの作成
腎細胞がんのデータにおいて、scSurvを空間トランスクリプトームデータに適用しました。その結果、組織切片上のどの領域が高い予後リスク(ハザード)を示すかを可視化する「ハザードマッピング」に成功しました。解析から、特定のT細胞が存在する領域が高いリスクと関連していることが明らかになりました。 -
感染症への応用
本手法はがんに限らず、COVID-19患者のデータにも適用可能であることが示されました。その解析により、特定の単球集団が重症化や入院期間の長期化に寄与していることが明らかとなり、疾患横断的な有用性が示されました。
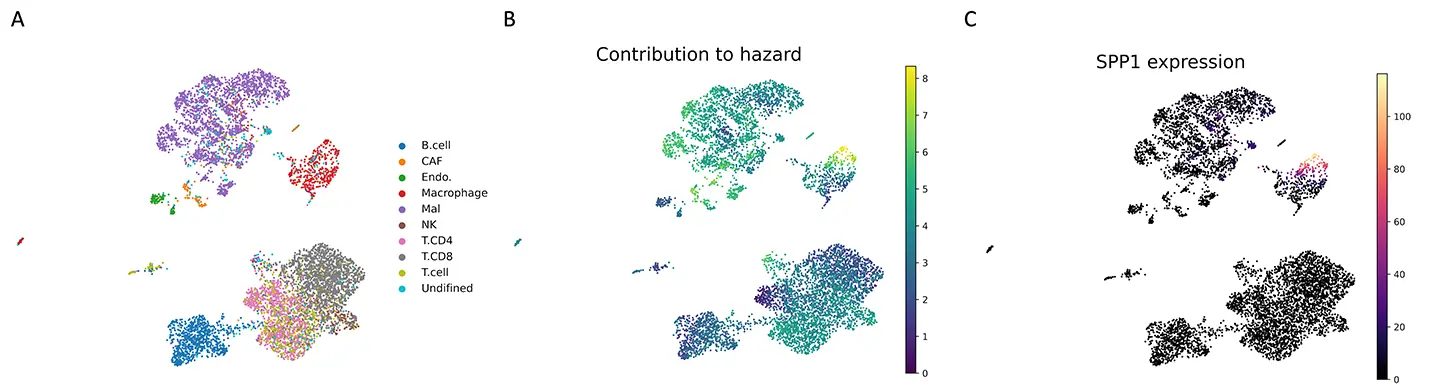
(A)メラノーマの細胞タイプ注釈。各点は各単一細胞を表している。
(B)scSurvで推定された各細胞のハザードへの寄与度。
(C)SPP1遺伝子の発現。scSurvで推定されたハザードへの寄与度が大きい(予後に悪い)細胞の分布と一致している。
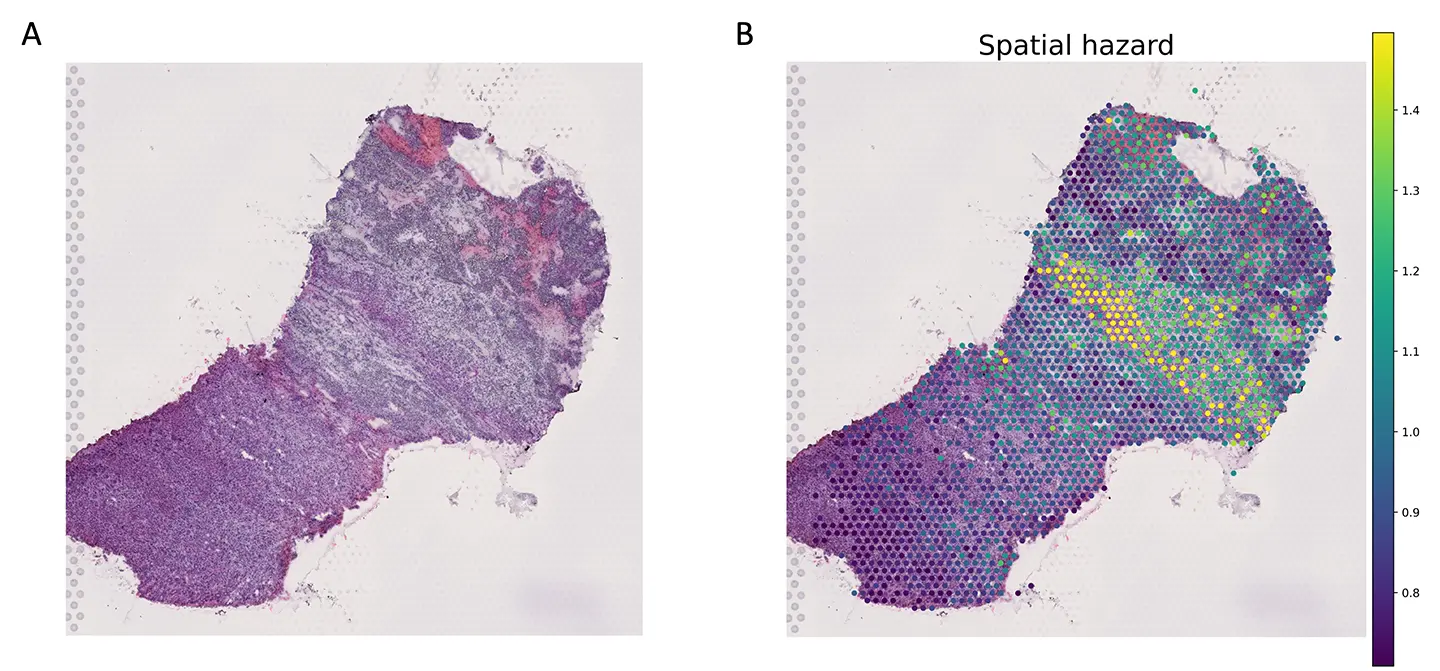
(A)腎細胞がんの病理切片のHE染色画像。
(B)scSurvにより空間ハザードを推定して病理切片上にマッピングしたもの。
社会的インパクト
本手法は、すでに世界中に蓄積されている膨大な「バルクRNAシーケンスデータ」と「臨床情報」を、最新の単一細胞解析の知見を用いて再解釈することを可能にします。これにより、新たな大規模な単一細胞RNAシーケンスデータを取得することなく、既存データから単一細胞解像度の知見を引き出すことができ、医学研究におけるコスト削減と研究効率の向上に大きく貢献します。
また、予後に悪影響を及ぼす具体的な細胞集団や分子メカニズムをピンポイントで特定できることから、新たな治療標的の発見や、患者ごとのリスクに応じた個別化医療の実現を加速させることが期待されます。
今後の展開
今後は本手法をさらに発展させ、特定の遺伝子変異の有無や薬剤投与履歴といった患者ごとの背景因子に応じて、各細胞の予後への寄与度がどのように変化するかを推定できるモデルへと改良を進めます。これにより、患者ごとの遺伝的背景や治療状況を踏まえた、より精密な予後予測が可能となります。
将来的には、こうした高解像度の情報を基に、最適な治療タイミングや薬剤選択を支援する次世代の個別化医療の実現に貢献することを目指します。
付記
本研究は、これらの事業からの支援を受けて行いました。
- 日本学術振興会(JSPS)(課題番号 22H04925、23H04938、23K16991)
- 日本医療研究開発機構(AMED)(課題番号 JP25gm2010002、JP25nk0101112、JP25wm0625519、JP25wm0325068、JP25zf0127012、JP25tm0424226、24ama221609h0001)
- 科学技術振興機構(JST) ムーンショット型研究開発事業(目標 2)(課題番号 JPMJMS2025)
- 国立がん研究センター研究開発費(課題番号 2024-A-6)
さらに、東京科学大学 高深度オミクス医学研究拠点整備事業からの支援を受けました。計算資源として、東京科学大学のスーパーコンピュータ TSUBAME3.0 および東京大学ヒトゲノム解析センターのスーパーコンピュータ SHIROKANE を利用しました。
用語説明
- [用語1]
- 単一細胞RNAシーケンス:組織を構成する細胞を一つずつ分離し、個々の細胞内の遺伝子の発現量を網羅的に解析することで、細胞ごとの機能や状態の違いを明らかにする技術。
- [用語2]
- 深層生成モデル:ディープラーニング(深層学習)を用いてデータが生成される背後の法則性を学習し、複雑なデータからの特徴抽出や、元のデータの特徴を捉えた新しいデータの生成を行うAIモデル。
- [用語3]
- Cox比例ハザードモデル:生存期間などの「イベント発生までの時間」を扱う生存解析において最も広く用いられる統計手法で、治療法や遺伝子発現などの要因がイベント発生リスク(ハザード)を何倍にするかを評価するモデル。
- [用語4]
- 空間トランスクリプトーム解析:組織切片上の位置情報と遺伝子発現情報を紐づけて解析することで、組織内のどの場所でどのような遺伝子が働いているかを地図のように可視化できる技術。
- [用語5]
- バルクRNAシーケンス:組織を細胞単位に分けず、塊のまま解析することで、そこに含まれる多数の細胞の平均的な遺伝子の働きを調べる、これまで広く用いられてきた標準的な解析手法。
論文情報
- 掲載誌:
- Bioinformatics
- タイトル:
- scSurv: a deep generative model for single-cell survival analysis
- 著者:
- Chikara Mizukoshi, Yasuhiro Kojima, Shuto Hayashi, Ko Abe, Daisuke Kasugai, Teppei Shimamura
研究者プロフィール
水越 周良 Chikara Mizukoshi
東京科学大学 総合研究院 難治疾患研究所 計算システム生物学分野 大学院生
研究分野:バイオインフォマティクス、データ科学、人工知能

小嶋 泰弘 Yasuhiro Kojima
国立がん研究センター研究所 計算生命科学ユニット 独立ユニット長
兼務:東京科学大学 総合研究院 難治疾患研究所 計算システム生物学分野 連携研究員
研究分野:バイオインフォマティクス、データ科学、人工知能

島村 徹平 Teppei Shimamura
東京科学大学 総合研究院 難治疾患研究所 計算システム生物学分野 教授
研究分野:システム生物学、バイオインフォマティクス、データ科学、人工知能
